


On November 30th science journalists were abuzz with news from the latest results from Deepmind, a wholly-owned subsidiary of Alphabet (FKA Google).
Deepmind had released their results from the latest round of CASP, the Critical Assessment of protein Structure Prediction. CASP is a biennial academic exercise where developers of protein structure prediction algorithms compete to produce the best model of a panel of proteins that experimentalists had that year, but crucially, hadn’t been released into the public domain. Deepmind’s Alphafold2 program won CASP14 (2020) and had done better than any program had done before, prompting some to proclaim that “the protein folding problem was solved”.
So, what is this problem, what have Deepmind done, and why you should approach the claims with some skepticism?
Proteins do almost everything inside you. As you read this article, light from the screen enters your eye. This light is focused by your retina – a lens, the shape of which is supported and altered by proteins. The light then interacts with proteins in the back of your eye. These proteins change shape in response to the light, and a signal is sent along your optic nerve where it is interpreted in your brain. The eyebrow you just raised after reading those sentences is made of and controlled by proteins.
DNA is the blueprint for proteins. The DNA sequence of a gene determines a protein’s sequence. The sequence of the protein determines its fold. The fold of the protein determines what it does and how it does it. The human genome encodes for at least 20,000 proteins (estimates vary depending upon your definition) and those proteins do much of the chemistry that makes you, you.
Proteins are assembled from a chemical alphabet of 20 amino acids. These amino acids have different properties and together will determine the fold and function of the protein. Glycine, for example is small and flexible – it will often be found in surface loops and flexible parts of the protein. Phenylalanine is large and hydrophobic (water-hating) and is often found in the core of a protein, hidden away from surrounding water. Glutamate is acidic, Arginine is basic, and so and so on.
How proteins fold and what they look like is a subject of intense study. We can solve the structure of proteins using a range of biophysical techniques – the most prolific of which is X-ray crystallography. Other techniques such as cryo-electron microscopy (currently undergoing a massive “resolution revolution”) and nuclear magnetic resonance (NMR) have different strengths that can be employed to solve different protein structures. These endeavors can be time consuming and expensive (my personal best is 4 weeks from “clone to structure” but typically a structural biology campaign lasts months or years), but to my mind there is no technique other than genetics that has contributed as much to our understanding of how life works than structural biology. Most protein structures determined are made publicly available, open to all in the protein data bank (PDB).
Structural biology is used not only to find out “how stuff works” but also “why stuff goes wrong” in the cases of diseases, and “can we fix the stuff that went wrong” in the fields of drug and therapeutic design. Pharmaceutical companies direct a significant amount of time, money, and effort into solving the structures of medically relevant proteins so that they can design drugs to treat various diseases, from cancers to microbial infection.
Protein structure prediction is what Alphafold is designed to do. The basic idea is that you feed the protein sequence (derived from information from the human genome project, for example) in at one end, and the structure pops out at the other. This isn’t new, but it is difficult. In 1969, Cyrus Levinthal presented a paper regarding a thought experiment about protein folding. If one considers a small, 100 amino acid protein, it will have 99 peptide bonds (bonds linking adjacent amino acids) and each of those bonds has 2 different angles and each angle has (roughly) 3 favourable conformations. Therefore the 100 amino acid protein would have 3198 different potential conformations. If the protein were to sequentially explore these conformations, even on a picosecond timescale, an individual protein molecule would take longer than the lifetime of the universe to sample them all. Your proteins routinely fold within microseconds or milliseconds of being produced. This disconnect is known as Levinthal’s paradox.
Alphafold2 is the latest iteration of Deepmind’s attempts at proteins structure prediction. In 2018, competing in CASP13, they came first with Alphafold. In 2018, the press announced that Deepmind had solved the protein folding problem. Now, in 2020, competing in CASP14 with Alphafold2, the press have announced that Deepmind have solved the protein folding problem. There is insufficient space to tell you how Alphafold2 works, but basically it uses deep neural networks trained on open protein structure and sequence databases. If you want more information, I can direct you to Deepmind’s blog and the Nature paper published after the CASP13.
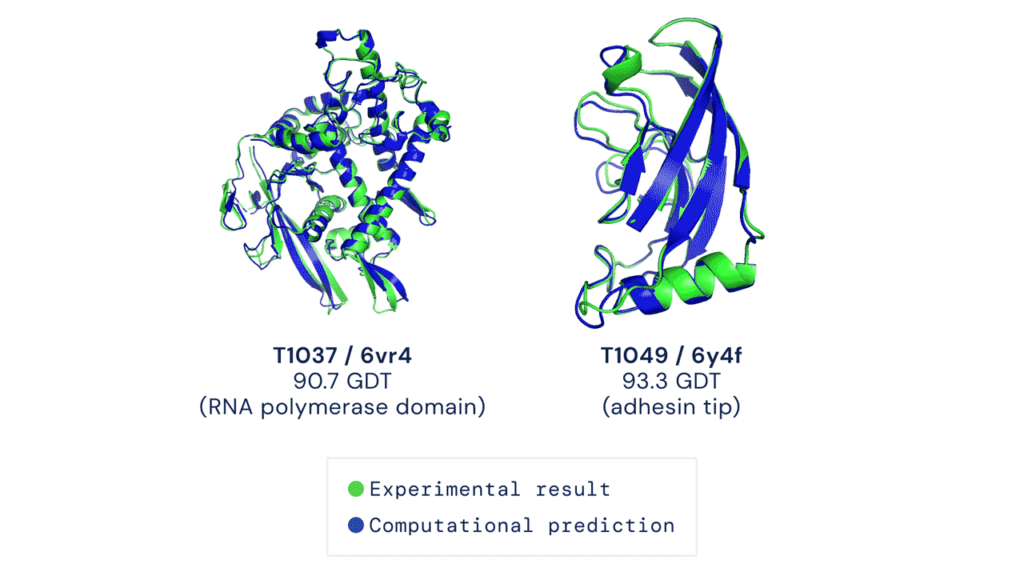
So, on the surface of it, this is a hugely promising development. Being able to accurately predict the structure of every protein would be transformative for both basic biology, but also biomedical research and potentially drug design. Why should we be skeptical? That Alphafold2 has obtained excellent results is not in question – the results have not been written up yet (but almost certainly will be), so all we have to go on right now is Deepmind’s CASP14 blog post and a couple of paragraphs in the CASP14 abstract book (PDF – Alphafold2 is on page 22), and they indeed suggest that Alphafold2 had achieved unparalleled accuracy. Conversations with colleagues associated with CASP suggest that Alphafold2 has obtained the best set of results yet – “often unimprovable” was one colleague’s assessment. However, it seems that the technique has still not reached the accuracy needed for drug design, or detailed analysis of enzyme mechanisms.
The potential problems around Alphafold2 stem from the problems met with Alphafold. Alphafold is not open source. There is no server that I can send my protein sequence to. There is no Github repository from where I can download the source code (There is a Github repository to rerun exactly what they did in CASP13, but crucially it is hobbled so that it may only work on the CASP13 targets, the experimentally determined structures for which are now freely available). There are reverse-engineered implementations such as ProSPr, which are similar but not identical.
There are two reasons I find this troubling: Firstly, is that Alphafold is closed, and yet it is built upon open databases (Uniprot, PDB) and uses academic open-source software (HHBlits, JackHmmr, OpenMM and the Amber forcefield for molecular modelling).
Secondly, we know that science moves fastest when methods, data, and information are shared. Open science means that everyone can peer-review everything. If we are granted a look behind the curtain, we can see how everything is put together. Those who are inclined can tinker with the nuts and bolts of Alphafold2 and maybe break it. Maybe make it better, or faster. Make it do a thing that no-one has envisaged would be do-able, or even useful. Maybe someone from an unrelated field will be inspired to take a piece of Alphafold2 and re-tool it for another task.

Unfortunately, Deepmind has form in the ‘keeping their toys to themselves’ game. AlphaGo was a program designed to play the strategy game, Go. It is the only program to beat a professional Dan-9 Go master. Despite widespread interest at the time, the source code was never released. Again, enthusiasts have had to reverse engineer the code from Deepmind’s descriptions. Keeping the knowledge behind winning at Go is one thing, but if Alphafold2 is as transformative as they claim, not releasing it is, to my mind, troubling and immoral. Medical advances could be made if this technology is as good as they claim. Lives could be saved or improved by information gleaned from the models that Alphafold2 produces.
I’d like to end this piece with a plea: if by any chance people from Deepmind/Alphafold are reading this, please make it open source or make a server where we can submit our sequences. I would think that the structural biology community would be extremely excited to try your new methods on our problems. Until you make your methods accessible and available, we will not be able to figure out how transformative your advances really are.